DeepMind and EMBL release the most complete database of predicted 3D structures of human proteins
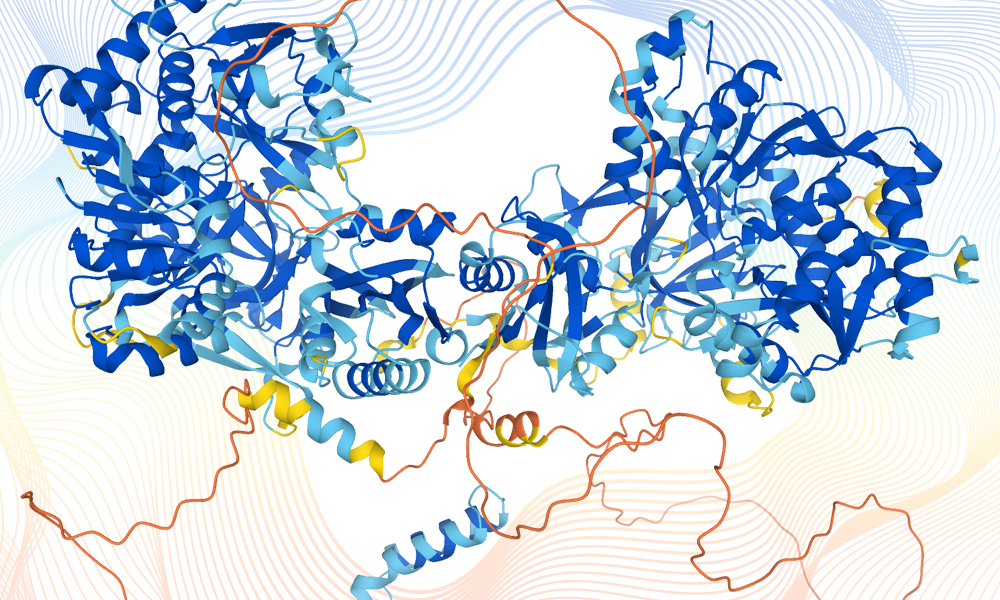
Protein structures representing the data obtained via AlphaFold. Source image: AlphaFold. Design credit: Karen Arnott/EMBL-EBI
DeepMind and EMBL release the most complete database of predicted 3D structures of human proteins
Protein structures representing the data obtained via AlphaFold. Source image: AlphaFold. Design credit: Karen Arnott/EMBL-EBI
DeepMind and EMBL release the most complete database of predicted 3D structures of human proteins
Vicky Hatch, EMBL
https://www.embl.org/news/science/alphafold-database-launch/
Partners use AlphaFold, the AI system recognised last year as a solution to the protein structure prediction problem, to release more than 350,000 protein structure predictions including the entire human proteome to the scientific community.
**
DeepMind today announced its partnership with the European Molecular Biology Laboratory (EMBL), Europe’s flagship laboratory for the life sciences, to make the most complete and accurate database yet of predicted protein structure models for the human proteome. This will cover all ~20,000 proteins expressed by the human genome, and the data will be freely and openly available to the scientific community. The database and artificial intelligence system provide structural biologists with powerful new tools for examining a protein’s three-dimensional structure, and offer a treasure trove of data that could unlock future advances and herald a new era for AI-enabled biology.
AlphaFold’s recognition in December 2020 by the organisers of the Critical Assessment of protein Structure Prediction (CASP) benchmark as a solution to the 50-year-old grand challenge of protein structure prediction was a stunning breakthrough for the field. The AlphaFold Protein Structure Database builds on this innovation and the discoveries of generations of scientists, from the early pioneers of protein imaging and crystallography, to the thousands of prediction specialists and structural biologists who’ve spent years experimenting with proteins since. The database dramatically expands the accumulated knowledge of protein structures, more than doubling the number of high-accuracy human protein structures available to researchers. Advancing the understanding of these building blocks of life, which underpin every biological process in every living thing, will help enable researchers across a huge variety of fields to accelerate their work.
Last week, the methodology behind the latest highly innovative version of AlphaFold, the sophisticated AI system announced last December that powers these structure predictions, and its open source code were published in Nature. Today’s announcement coincides with a second Nature paper that provides the fullest picture of proteins that make up the human proteome, and the release of 20 additional organisms that are important for biological research.
“Our goal at DeepMind has always been to build AI and then use it as a tool to help accelerate the pace of scientific discovery itself, thereby advancing our understanding of the world around us,” said DeepMind Founder and CEO Demis Hassabis, PhD. “We used AlphaFold to generate the most complete and accurate picture of the human proteome. We believe this represents the most significant contribution AI has made to advancing scientific knowledge to date, and is a great illustration of the sorts of benefits AI can bring to society.”
AlphaFold is already helping scientists to accelerate discovery
The ability to predict a protein’s shape computationally from its amino acid sequence – rather than determining it experimentally through years of painstaking, laborious and often costly techniques – is already helping scientists to achieve in months what previously took years.
“The AlphaFold database is a perfect example of the virtuous circle of open science,” said EMBL Director General Edith Heard. “AlphaFold was trained using data from public resources built by the scientific community so it makes sense for its predictions to be public. Sharing AlphaFold predictions openly and freely will empower researchers everywhere to gain new insights and drive discovery. I believe that AlphaFold is truly a revolution for the life sciences, just as genomics was several decades ago and I am very proud that EMBL has been able to help DeepMind in enabling open access to this remarkable resource.”
AlphaFold is already being used by partners such as the Drugs for Neglected Diseases Initiative (DNDi), which has advanced their research into life-saving cures for diseases that disproportionately affect the poorer parts of the world, and the Centre for Enzyme Innovation (CEI) is using AlphaFold to help engineer faster enzymes for recycling some of our most polluting single-use plastics. For those scientists who rely on experimental protein structure determination, AlphaFold’s predictions have helped accelerate their research. For example, a team at the University of Colorado Boulder is finding promise in using AlphaFold predictions to study antibiotic resistance, while a group at the University of California San Francisco has used them to increase their understanding of SARS-CoV-2 biology.
The AlphaFold Protein Structure Database
The AlphaFold Protein Structure Database builds on many contributions from the international scientific community, as well as AlphaFold’s sophisticated algorithmic innovations and EMBL-EBI’s decades of experience in sharing the world’s biological data. DeepMind and EMBL’s European Bioinformatics Institute (EMBL-EBI) are providing access to AlphaFold’s predictions so that others can use the system as a tool to enable and accelerate research and open up completely new avenues of scientific discovery.
“This will be one of the most important datasets since the mapping of the Human Genome,” said EMBL Deputy Director General, and EMBL-EBI Director Ewan Birney. “Making AlphaFold predictions accessible to the international scientific community opens up so many new research avenues, from neglected diseases to new enzymes for biotechnology and everything in between. This is a great new scientific tool, which complements existing technologies, and will allow us to push the boundaries of our understanding of the world.”
In addition to the human proteome, the database launches with ~350,000 structures including 20 biologically-significant organisms such as E.coli, fruit fly, mouse, zebrafish, malaria parasite and tuberculosis bacteria. Research into these organisms has been the subject of countless research papers and numerous major breakthroughs. These structures will enable researchers across a huge variety of fields – from neuroscience to medicine – to accelerate their work.
The future of AlphaFold
The database and system will be periodically updated as we continue to invest in future improvements to AlphaFold, and over the coming months we plan to vastly expand the coverage to almost every sequenced protein known to science – over 100 million structures covering most of the UniProt reference database.
To learn more, please see the Nature papers describing our full method and the human proteome, and read the Authors’ Notes. See the open-source code to AlphaFold if you want to view the workings of the system, and Colab notebook to run individual sequences. To explore the structures, visit EMBL-EBI’s searchable database that is open and free to all.
AlphaFold
Protein Structure Database
Developed by DeepMind and EMBL-EBI
https://alphafold.ebi.ac.uk/
AlphaFold DB provides open access to protein structure predictions for the human proteome and 20 other key organisms to accelerate scientific research.
**
Background
AlphaFold is an AI system developed by DeepMind that predicts a protein’s 3D structure from its amino acid sequence. It regularly achieves accuracy competitive with experiment.
DeepMind and EMBL’s European Bioinformatics Institute (EMBL-EBI) have partnered to create AlphaFold DB to make these predictions freely available to the scientific community. The first release covers the human proteome and the proteomes of several other key organisms. In the coming months we plan to expand the database to cover a large proportion of all catalogued proteins (the over 100 million in UniRef90).
Q8I3H7: May protect the malaria parasite against attack by the immune system. Mean pLDDT 85.57.
In CASP14, AlphaFold was the top-ranked protein structure prediction method by a large margin, producing predictions with high accuracy. While the system still has some limitations, the CASP results suggest AlphaFold has immediate potential to help us understand the structure of proteins and advance biological research.
Q8W3K0: A potential plant disease resistance protein. Mean pLDDT 82.24.
What’s next?
AlphaFold DB will continue to expand in the coming weeks and months, so if you can’t find what you’re looking for right now, please follow DeepMind and EMBL-EBI’s social channels for updates. In the meantime the AlphaFold source code and Colab notebook can be used to predict the structures of proteins not yet in AlphaFold DB.
AIが予測したヒトタンパク質の画期的データベース、オンラインで公開
AFPBB News
https://www.afpbb.com/articles/-/3358076?cx_part=search
生体を構成する主成分であるタンパク質の構造をAI(人工知能)に学習・予測させたこれまでで最も網羅的なデータベースが先週、オンライン上で公開された。画期的な成果は「生物学研究を根本から変える」と注目を集めている。
公開されたのは、ヒトのゲノム(全遺伝情報)によって発現するタンパク質、約2万種類のデータベース。米IT大手グーグル(Google)の親会社アルファベット(Alphabet)傘下のAI開発企業ディープマインド(DeepMind)と、欧州分子生物学研究所(EMBL)が無料で一般公開した。
生物の細胞はどれもタンパク質が絶え間なく発する指令によって、健康を維持したり、感染を防いだりするための機能を発揮させる。
ゲノムとは異なり、プロテオーム(発現し得る全タンパク質)は遺伝的指令や環境の刺激によって常に変化している。
タンパク質が細胞内でどのように作用するか、つまり、「折りたたみ」と呼ばれる立体構造のどの形に落ち着くかを理解する研究は、何十年にもわたって科学者たちを魅了してきた。
しかし、それぞれのタンパク質の正確な機能を実験によって直接明らかにすることは非常に困難で、過去50年間で、ヒトのプロテオームのアミノ酸のうちわずか17%しか明らかになっていない。アミノ酸はタンパク質の構成成分。
今回のデータベースを構築するために科学者らは、アミノ酸配列に基づいてタンパク質の形状を正確に予測する最新の機械学習プログラムを使用。既知のタンパク質構造17万種類のデータベースをAI「アルファフォールド(AlphaFold)」に学習させ、人間で発現し得る全タンパク質のうち58%の構造を予測した。
遺伝性疾患や抗菌剤耐性の研究など、応用できる可能性は極めて大きいと期待される。
Protein structure
Wikipedia
https://en.wikipedia.org/wiki/Protein_structure
Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. Proteins are polymers – specifically polypeptides – formed from sequences of amino acids, the monomers of the polymer. A single amino acid monomer may also be called a residue indicating a repeating unit of a polymer. Proteins form by amino acids undergoing condensation reactions, in which the amino acids lose one water molecule per reaction in order to attach to one another with a peptide bond. By convention, a chain under 30 amino acids is often identified as a peptide, rather than a protein. To be able to perform their biological function, proteins fold into one or more specific spatial conformations driven by a number of non-covalent interactions such as hydrogen bonding, ionic interactions, Van der Waals forces, and hydrophobic packing. To understand the functions of proteins at a molecular level, it is often necessary to determine their three-dimensional structure. This is the topic of the scientific field of structural biology, which employs techniques such as X-ray crystallography, NMR spectroscopy, cryo electron microscopy (cryo-EM) and dual polarisation interferometry to determine the structure of proteins.
Protein structures range in size from tens to several thousand amino acids. By physical size, proteins are classified as nanoparticles, between 1–100 nm. Very large protein complexes can be formed from protein subunits. For example, many thousands of actin molecules assemble into a microfilament.
A protein usually undergoes reversible structural changes in performing its biological function. The alternative structures of the same protein are referred to as different conformations, and transitions between them are called conformational changes.
**
タンパク質構造
ウィキペディア(Wikipedia)
https://ja.wikipedia.org/wiki/タンパク質構造
タンパク質構造(Protein structure)では、タンパク質の構造について記す。タンパク質は全ての生物が持つ、重要な生体高分子の1つである。タンパク質は炭素、水素、窒素、リン、酸素、硫黄の原子から構成された、残基と言われるアミノ酸のポリマーである。ポリペプチドとも呼ばれるこのポリマーは20種類のL-α-アミノ酸の配列からできている。40以下のアミノ酸から構成されるものは、しばしばタンパク質ではなくペプチドと呼ばれる。その機能を発現するために、タンパク質は水素結合、イオン結合、ファンデルワールス力、疎水結合などの力によって、特有のコンフォメーションをとるように折り畳まれる。分子レベルのタンパク質の機能を理解するには、その三次元構造を明らかにしなければならない。これは構造生物学の研究分野で、X線回折や核磁気共鳴分光法などの技術が使われる。
アミノ酸残基の数は特定の生化学的機能を果たす際に重要で、機能を持ったドメインのサイズとしては40から50残基が下限となる。タンパク質自体の大きさはこの下限から数1000残基のものまで様々で、その平均は約300残基と見積もられている。多くのG-アクチンがアクチン繊維(F-アクチン)を作るように、多くのタンパク質サブユニットが集合して1つの構造を作ることもある。